내생성: 베이지안 분석
불확실을 감안한 분석
set.seed(1)
N <- 1000
gene <- rnorm(N)
power <- gene + (e1 = rnorm(N))
height <- 180 + 5*gene + (e2 = rnorm(N))
ability <- power + height/10 + (e3 = rnorm(N))
summary(lm(ability ~ height)) # 예측 모형
## ## Call: ## lm(formula = ability ~ height) ## ## Residuals: ## Min 1Q Median 3Q Max ## -5.1616 -1.0461 -0.0145 1.0365 5.0071 ## ## Coefficients: ## Estimate Std. Error t value Pr(>|t|) ## (Intercept) -35.56398 1.59503 -22.30 <2e-16 *** ## height 0.29756 0.00886 33.59 <2e-16 *** ## --- ## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 ## ## Residual standard error: 1.491 on 998 degrees of freedom ## Multiple R-squared: 0.5306, Adjusted R-squared: 0.5301 ## F-statistic: 1128 on 1 and 998 DF, p-value: < 2.2e-16
summary(lm(ability ~ height + power)) # 원인 결과 모형
## ## Call: ## lm(formula = ability ~ height + power) ## ## Residuals: ## Min 1Q Median 3Q Max ## -3.2322 -0.7124 -0.0099 0.7173 3.0623 ## ## Coefficients: ## Estimate Std. Error t value Pr(>|t|) ## (Intercept) -0.372789 1.551017 -0.24 0.81 ## height 0.102167 0.008614 11.86 <2e-16 *** ## power 1.013926 0.031168 32.53 <2e-16 *** ## --- ## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 ## ## Residual standard error: 1.039 on 997 degrees of freedom ## Multiple R-squared: 0.7723, Adjusted R-squared: 0.7718 ## F-statistic: 1691 on 2 and 997 DF, p-value: < 2.2e-16
cov(height, power + e3)
## [1] 5.604122
앞의 예에서 \(\mathbb{C}\textrm{ov}(x, e)\) 을 정확하게 모른다면, 여러 가지 가능한 값을 대입한 후, 인과관계 계수를 살펴볼 수 있을 것이다.[1]
[1] : 흔히 이렇게 가정이 달라짐에 따라 분석 결과 어떻게 달라지는지를 확인하는 과정을 민감도 분석(Sensitivity Analysis)라고 한다.
\(\mathbb{C}\textrm{ov}(x, e)\) 가 4 또는 5 또는 6일지 모른다면,
library(lavaan)
causalModel <- '
ability ~ height
ability ~~ b4*height
b4 == 4'
dat = data.frame(gene=gene, power=power, height=height, ability=ability)
fit <- sem(causalModel, data=dat)
coef(fit)
## ability~height b4 ability~~ability height~~height ## 0.156 4.000 2.784 28.338
causalModel <- '
ability ~ height
ability ~~ b4*height
b4 == 5'
dat = data.frame(gene=gene, power=power, height=height, ability=ability)
fit <- sem(causalModel, data=dat)
#fit <- sem(causalModel, data=dat, estimator='GLS')
coef(fit)
## ability~height b4 ability~~ability height~~height ## 0.121 5.000 3.102 28.338
causalModel <- '
ability ~ height
ability ~~ b4*height
b4 == 6'
dat = data.frame(gene=gene, power=power, height=height, ability=ability)
#fit <- sem(causalModel, data=dat)
fit <- sem(causalModel, data=dat, estimator='GLS')
#fit <- sem(causalModel, data=dat, estimator='WLS')
coef(fit)
## ability~height b4 ability~~ability height~~height ## 0.086 6.000 3.491 28.366
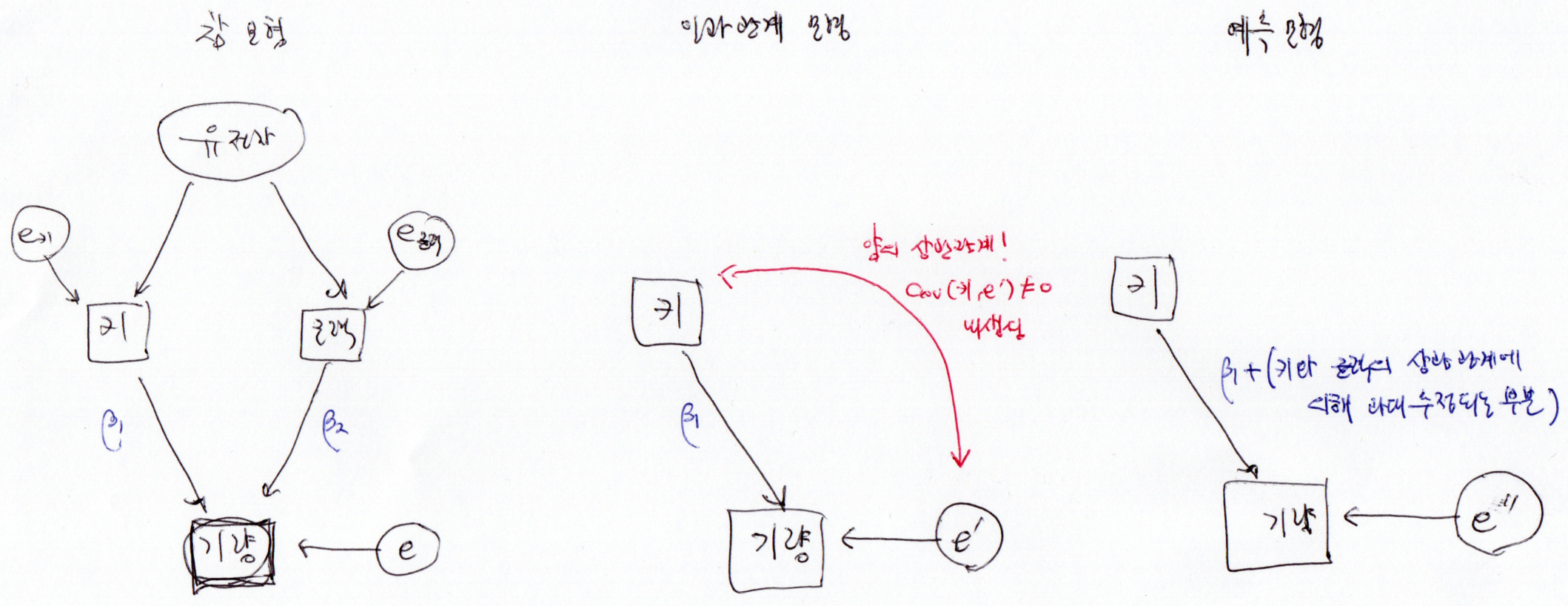
인과관계 계수는 0.15 또는 0.12 또는 0.09로 추정되었다. 그런데 이렇게만 말하고 끝내기가 꺼림칙한데 왜냐하면 이 추정값에는 표준오차도 있기 때문이다.
베이지안 분석은 모형이 존재한다면, 모수의 불확실성을 감안하여 분석을 진행할 수 있다.
베이지안 분석
예를 들어 \(\mathbb{C}\textrm{ov}(x, e)\) 의 정확한 값을 알지 못하지만, 그렇다고 아예 어떤 값인지 모르는 것도 아니라면 어떨까? 이를테면 5를 중심으로 그 근처일 가능성이 크고, 이를 확률분포로 평균 5이고, 표준편차 1인 정규분포로 나타낼 수 있다고 하자.
그리고 오차 \(e\) 는 \(x\) 에 따른 조건부 평균이 \(\rho (x-\bar{x})\) 이고, 분산이 \(1/\tau^2\) 인 정규분포를 띈다고 가정해보자. \(e\) 의 분포는 다음과 같이 나타낼 수 있다. ( \(\mathbb{C}\textrm{ov}(x, e)\) 이 양이고, \(x\) 와 \(e\) 가 선형적 관계를 가정했다.)
\(e|x \sim \mathcal{N}(\rho \cdot (x-\bar{x}), 1/\tau^2)\)
전체 모형은 다음과 같다.
\[y = \beta_0 + \beta_1 x + e, \ \ e \sim \mathcal{N}(\rho \cdot (x-\bar{x}), 1/\tau^2)\]
(이때 \(\mathbb{V}ar(e) = \rho^2 \mathbb{V}\textrm{ar}(x) + 1/\tau^2, \mathbb{C}\textrm{ov}(x,e) = \rho\mathbb{V}\textrm{ar}(x)\) 이 된다.)
여기서 \(x\) 를 주어진 값으로 생각하면, \(\mathbb{E}(x)\) 와 \(\mathbb{V}\textrm{ar}(x)\) 역시 주어진 \(x\) 에서 계산할 수 있는 상수이고, \(\rho\) 는 \(\mathbb{C}\textrm{ov}(x,e)/\mathbb{V}\textrm{ar}(x)\) 으로 구할 수 있다.
자료는 \(x\) 와 \(y\) (키와 기량)이고 모수는 \(\beta_0, \beta_1, \sigma^2 = \tau^2, \mathbb{C}\textrm{ov}(e,x)\) 이고, 이들 모수를 통해 \(\rho = \mathbb{C}\textrm{ov}(x,e)/\mathbb{V}\textrm{ar}(x), \mathbb{V}ar(e) = \rho^2 \mathbb{V}\textrm{ar}(x) + 1/\tau^2\) 을 구할 수 있다.
이 모형을 JAGS를 활용하여 분석해보자.
JAGS
JAGS는 Just Another Gibbs Sampler의 약자로, 다음의 링크에서 다운 받을 수 있다.
JAGS는 이름에서 알 수 있듯이 모수의 사전분포, 모형, 그리고 자료를 통해 사후분포를 깁스 샘플링(Gibbs sampling)으로 알아낸다.
다음의 model은 위의 모형을 JAGS 언어로 나타낸 것이다. 사전 분포를 다음과 같이 가정했다. (JAGS의 dnorm은 \(\tau=1/\sigma^2\) 로 정규분포를 나타냄을 유의하자.)
\(\beta_0 \sim \mathcal{N}(0, 10^2)\)
\(\beta_1 \sim \mathcal{N}(0, 10^2)\)
\(\mathbb{C}\textrm{ov}(x,e) \sim \mathcal{N}(5, 1^2)\)
\(\sigma^2 \sim \textrm{Unif}(0,100)\)
model <- '
# Simple linear regression with endogeneity
model {
# model
for (i in 1:N) {
meany[i] <- beta0 + xx[i]*beta1
meane[i] <- rho*(xx[i]-meanxx)
meanype[i] <- meany[i] + meane[i]
y[i] ~ dnorm(meanype[i], tau)
}
# priors
rho <- covXE/varX
tau <- pow(sigma2, -2)
varE <- pow(rho, 2)*varX + pow(sigma2,2)
covXE ~ dnorm(5, 1)
beta0 ~ dnorm(0, 0.01)
beta1 ~ dnorm(0, 0.01)
sigma2 ~ dunif(0,100)
}
'
cat(model, file="currentModel.j")
이제 일반적인 변수이름으로 되어 있는 독립변수 x, 결과변수 y에 데이터를 넣어준 후, jags.model, update, 그리고 coda.samples로 MCMC(Markov Chain Monte Carlo)를 돌린다.
x = height
y = ability
require(rjags)
myj <- jags.model('currentModel.j',
data = list('xx'=x,
'meanxx' = mean(x),
'y'=y,
'N'=N,
varX = (N-1)*var(x)/N),
n.chains = 1)
update(myj, n.iter = 1000)
n.thin=1
mcmcsamp <- coda.samples(myj, c('beta1', 'covXE', 'varE'), 5000*n.thin, thin=n.thin)
summary(mcmcsamp)
## ## Iterations = 2001:7000 ## Thinning interval = 1 ## Number of chains = 1 ## Sample size per chain = 5000 ## ## 1. Empirical mean and standard deviation for each variable, ## plus standard error of the mean: ## ## Mean SD Naive SE Time-series SE ## beta1 0.05374 0.009578 0.0001355 0.005762 ## covXE 6.78595 0.353418 0.0049981 0.103474 ## varE 3.86127 0.196653 0.0027811 0.041798 ## ## 2. Quantiles for each variable: ## ## 2.5% 25% 50% 75% 97.5% ## beta1 0.03512 0.04546 0.05729 0.06152 0.0669 ## covXE 6.14288 6.52862 6.76884 7.02841 7.4936 ## varE 3.50240 3.72118 3.85434 3.99156 4.2693
plot(mcmcsamp)

안타깝게도 자기상관(autocorrelation)이 너무 커보인다. 자기상관을 없애는 방법은 thinning이 아니라 reparameterization이라고 한다.[참고]
간단하게 x, y 모두 평균 중심화를 하면, 다음과 같다.
# model
model <- '
# Simple linear regression with endogeneity
# x, y is centered
model {
# model
for (i in 1:N) {
meany[i] <- xx[i]*beta1
meane[i] <- rho*(xx[i]-meanxx)
meanype[i] <- meany[i] + meane[i]
y[i] ~ dnorm(meanype[i], tau)
}
# priors
rho <- covXE/varX
tau <- pow(sigma2, -2)
varE <- pow(rho, 2)*varX + pow(sigma2,2)
covXE ~ dnorm(5, 1)
#beta0 ~ dnorm(0, 0.01)
beta1 ~ dnorm(0, 0.01)
sigma2 ~ dunif(0,100)
}
'
cat(model, file="currentModel.j")
x = height
y = ability
require(rjags)
myj <- jags.model('currentModel.j',
data = list('xx'=x-mean(x),
'meanxx' = 0,
'y'=y-mean(y),
'N'=N,
varX = (N-1)*var(x)/N),
n.chains = 1)
update(myj, n.iter = 1000)
n.thin=1
mcmcsamp <- coda.samples(myj, c('beta1', 'covXE', 'varE'), 5000*n.thin, thin=n.thin)
summary(mcmcsamp)
## ## Iterations = 2001:7000 ## Thinning interval = 1 ## Number of chains = 1 ## Sample size per chain = 5000 ## ## 1. Empirical mean and standard deviation for each variable, ## plus standard error of the mean: ## ## Mean SD Naive SE Time-series SE ## beta1 0.1209 0.03484 0.0004927 0.002667 ## covXE 5.0058 0.95633 0.0135246 0.072968 ## varE 3.1465 0.35562 0.0050292 0.026203 ## ## 2. Quantiles for each variable: ## ## 2.5% 25% 50% 75% 97.5% ## beta1 0.05351 0.09754 0.1203 0.1448 0.1893 ## covXE 3.12756 4.35544 5.0197 5.6537 6.8537 ## varE 2.53294 2.89782 3.1175 3.3745 3.9057
plot(mcmcsamp)

만약 \(\mathbb{C}\textrm{ov}(x,e)\) 를 정말 잘 모르겠다면, 불확실성을 감안하여 사전분포를 다음과 같이 바꿀 수 있다.
\(\mathbb{C}\textrm{ov}(x,e) \sim \mathcal{N}(5, 10)\)
# model
model <- '
# Simple linear regression with endogeneity
# x, y is centered
# cov(X,e) is uncertain
model {
# model
for (i in 1:N) {
meany[i] <- xx[i]*beta1
meane[i] <- rho*(xx[i]-meanxx)
meanype[i] <- meany[i] + meane[i]
y[i] ~ dnorm(meanype[i], tau)
}
# priors
rho <- covXE/varX
tau <- pow(sigma2, -2)
varE <- pow(rho, 2)*varX + pow(sigma2,2)
covXE ~ dnorm(5, 0.1)
#beta0 ~ dnorm(0, 0.01)
beta1 ~ dnorm(0, 0.01)
sigma2 ~ dunif(0,100)
}
'
cat(model, file="currentModel.j")
x = height
y = ability
require(rjags)
myj <- jags.model('currentModel.j',
data = list('xx'=x-mean(x),
'meanxx' = 0,
'y'=y-mean(y),
'N'=N,
varX = (N-1)*var(x)/N),
n.chains = 3)
update(myj, n.iter = 1000)
n.thin=3
mcmcsamp <- coda.samples(myj, c('beta1', 'covXE', 'varE'), 5000*n.thin, thin=n.thin)
summary(mcmcsamp)
## ## Iterations = 2003:17000 ## Thinning interval = 3 ## Number of chains = 3 ## Sample size per chain = 5000 ## ## 1. Empirical mean and standard deviation for each variable, ## plus standard error of the mean: ## ## Mean SD Naive SE Time-series SE ## beta1 0.1413 0.1193 0.0009744 0.01044 ## covXE 4.4259 3.3700 0.0275156 0.29359 ## varE 3.3206 1.2815 0.0104630 0.10860 ## ## 2. Quantiles for each variable: ## ## 2.5% 25% 50% 75% 97.5% ## beta1 -0.1019 0.06263 0.146 0.2247 0.3651 ## covXE -1.9600 2.08371 4.278 6.6562 11.2879 ## varE 2.1494 2.42355 2.886 3.7834 6.7221
plot(mcmcsamp)

다음의 그림에서 \(\beta_1\) 과 \(\mathbb{C}\textrm{ov}(x,e)\) 의 결합확률밀도를 보면 \(\beta_1\) 의 불확실성은 \(\mathbb{C}\textrm{ov}(x,e)\) 에서 기인함을 확인할 수 있다.(그리고 깁스 샘플링인 것을 감안하면, \(\beta_1\) 샘플링의 자기상관이 높은 이유도 분명해보인다.)
str(mcmcsamp)
## List of 3 ## $ : 'mcmc' num [1:5000, 1:3] 0.0471 0.0477 0.0374 0.0592 0.0425 ... ## ..- attr(*, "dimnames")=List of 2 ## .. ..$ : NULL ## .. ..$ : chr [1:3] "beta1" "covXE" "varE" ## ..- attr(*, "mcpar")= num [1:3] 2003 17000 3 ## $ : 'mcmc' num [1:5000, 1:3] -0.0658 -0.0605 -0.0283 -0.0462 -0.0272 ... ## ..- attr(*, "dimnames")=List of 2 ## .. ..$ : NULL ## .. ..$ : chr [1:3] "beta1" "covXE" "varE" ## ..- attr(*, "mcpar")= num [1:3] 2003 17000 3 ## $ : 'mcmc' num [1:5000, 1:3] 0.118 0.15 0.134 0.128 0.135 ... ## ..- attr(*, "dimnames")=List of 2 ## .. ..$ : NULL ## .. ..$ : chr [1:3] "beta1" "covXE" "varE" ## ..- attr(*, "mcpar")= num [1:3] 2003 17000 3 ## - attr(*, "class")= chr "mcmc.list"
#str(mcmcsamp[[1]])
dat <- as.data.frame(mcmcsamp[[1]])
require(ggplot2)
#ggplot(dat, aes(x=beta1, y=covXE)) + geom_point(alpha=0.2) + geom_density2d()
ggplot(dat, aes(x=beta1, y=covXE)) + geom_point(alpha=0.1)

베이지안 분석의 단점
하지만 \(e\) 에 대한 정확한 모형이 없다면?
위에서 우리는 내생성이 존재하고, \(\mathbb{E}[e | x]\) 는 $x$에 비례하는 가장 간단한 모형을 가정했다.
\[e \sim \mathcal{N}(\rho \cdot (x-\bar{x}), 1/\tau^2)\]
하지만 \(e\) 에 대한 구체적인 분포를 알 수 없다면 베이지안 방법을 쓰기 어렵다.
이 때에는 GMM(Generalized Method of Moments)을 쓸 수 있다.
정리
여기서는 경로분석을 베이지안으로 구현해 보았다. (사실 회귀선은 하나뿐이었지만, 여럿이 존재해도 동일한 방식으로 구현할 수 있다.) 사실 베이지안 분석이란 게 크게 어려운 것이 없다. 간단하게 한 줄로 정리하자면,
모수의 사전 분포가 있고, 모형이 있고( \(\mathbb{P}(\vec{y} | \vec{\theta}))\), \(y\) : 관찰값, \(\theta\) : 모수), 관찰값이 있을 때, \(\mathbb{P}(\vec{\theta} | \vec{y})\) (모수의 사후 분포)를 구하는 것이다.
물론 이 사후 분포를 공식으로 계산하기 힘들기 때문에 샘플링을 활용한다. 여기서는 JAGS를 활용하여 샘플링하는 방법을 보였다. 샘플링은 계산부하가 많이 들어가는 과정이다. 이때 문제가 생기기도 한다. 위의 예에서는 \(\beta_1\) 의 샘플링이 자기 상관이 매우 커서, 재모수화( \(x\) , \(y\) 평균 중심화)로 해결했다.
마지막으로 문제 하나.
- 위의 예에서
N <- 100로 수정한 후 분석을 시도해보자. 오류가 발생한다면 어떻게 수정할 수 있는가?
Leave a comment